Editor's note: Learn more about cathodic protection for steel structures buried in soil in this new Materials Performance quarterly special feature, “The Science Behind It.” After you’ve read the MP article about the effect of pH and degree of environmental aeration on the corrosion rate, explore the science behind the corrosion problem, which is presented in several related CORROSION articles listed at the end of this article.
The conventional understanding of the cathodic protection (CP) mechanism is founded in the results of experiments carried out by Mears and Brown in 1938.1 They stated that, “For CP to be entirely effective, the local cathodes on the corroding specimen must be polarized to the potential of the unpolarized local anodes.” At that point, as illustrated in Figure 1, the corrosion current density (CD) is reduced to the exchange CD (i0) at which point the corrosion CD is zero.
This theory was supported by Dexter, et al.,2 who conducted polarization tests on steel in seawater and concluded that, “CP was achieved by polarizing to, or at least toward, the potential of the local action anodes in agreement with the Mears and Brown and Hoar theories.”
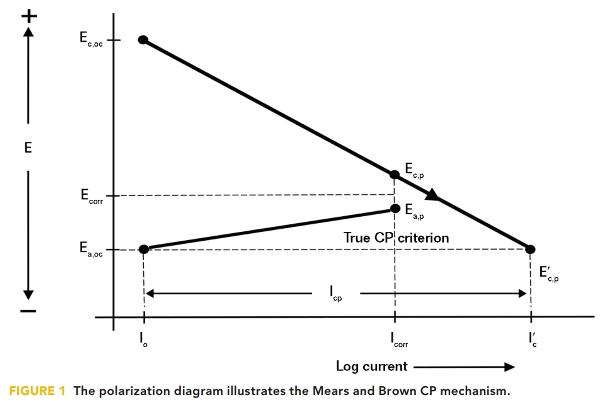
For complete CP protection, therefore, the true criterion for any corrosion cell is the open circuit potential of the corrosion cell anode (Ea,oc). Extending this electrical analogy to a structure having many corrosion cells means that for complete CP, all cathode sites need to be polarized electronegatively to the most electronegative anode site on the structure. However, this presents a serious problem because of the impracticality of determining the potential of the most electronegative open circuit anode on the structure.
Kuhn3 addressed this problem based on empirical data that were obtained on the CP of cast iron water mains in New Orleans, Louisiana, USA. He proposed a potential of –850 mV vs. copper/copper sulfate (Cu/CuSO4) electrode (CSE) “to which a pipe must be lowered in order to stop corrosion.” Although there was no verification on the effectiveness of this criterion at the time, it was assumed that this potential criterion was more electronegative than any open circuit anode potential on a ferrous structure.
The effectiveness of CP was further investigated by Schwerdtfeger and McDorman at the National Bureau of Standards4 in the early 1950s. The potential of steel electrodes, placed in soil samples of varying pH, were measured and plotted against pH, as illustrated in Figure 2.
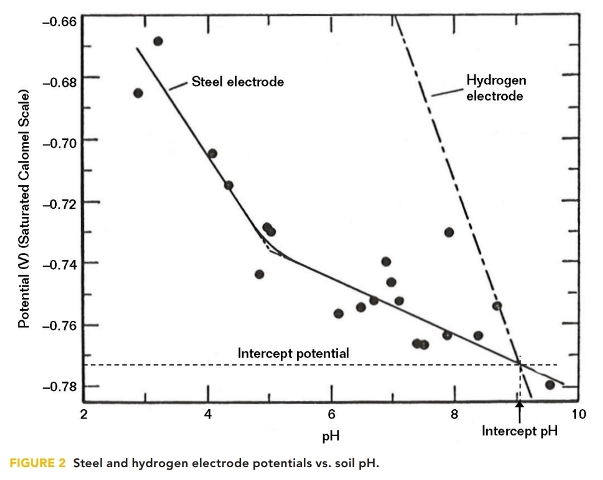
The soil samples, which were collected from different geographical locations across the United States, were intentionally deaerated so that the only possible cathodic reaction on the steel electrode would be the reduction of hydrogen ions (i.e., H+ + e– = Ho). The potential of a hydrogen electrode is thermodynamically related to pH. This relationship is plotted on Figure 2 along with the steel electrode corrosion potential.
For a steel electrode in a deaerated environment, the corrosion cell cathode potential would be the hydrogen electrode potential and the steel potential is assumed to be the anode potential of the corrosion cell. The potential difference between the steel coupon and the hydrogen electrode is considered the corrosion cell driving potential, which gradually diminishes as the soil pH increases and reaches zero at a potential of ~–770 mV vs. CSE, at which point the corrosion current would be expected to be zero. This point equates to a potential of –845 mV vs. CSE and occurs at ~pH 9. Besides providing theoretical validity to the –850 mV vs. CSE criterion, the results also suggest that increasing the pH would result in corrosion reduction.
The Role of pH in Controlling Steel Corrosion
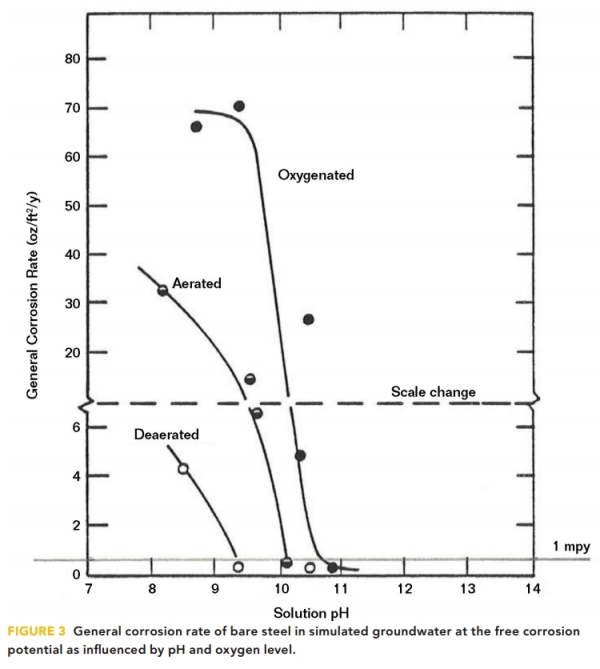
The effect of pH on the reduction of the steel corrosion rate was demonstrated in 1983 by Barlo, et al.5 for aerated, oxygenated, and deaerated conditions, as shown in Figure 3.
As the pH increases from pH 8, it is apparent that steel corrosion rates diminish to <1 mpy at pH 9.3 in deaerated conditions, at pH 10 for aerated conditions, and at pH 10.7 for oxygenated conditions.
Furthermore, increasing the environmental pH has been shown to be effective in reducing other forms of corrosion, such as stress corrosion cracking and sulfate-reducing bacteria.6-7
Cathodic Protection and pH
When CP current is applied to a structure, the current is transferred across the structure/electrolyte boundary by one or more of the following reduction reactions, depending on the environment and the magnitude of the CD. The common reduction reactions are shown in Equations (1) through (3):
H+ + e– → Ho (1)
• Hydrogen ion reduction in unaerated or low pH environments
2H2O + O2 + 4e– → 4OH– (2)
• Dissolved oxygen (DO) reduction in an aerated environment
2H2O + 2e– → H2 + 2OH– (3)
• Electrolysis of water
All of the foregoing reduction reactions result in an increase in the concentration of OH- ions at the structure/electrolyte interface and a proportionate increase in pH. Büchler,8 in 2013, illustrated this increase in pH on a Pourbaix diagram, as shown in Figure 4.
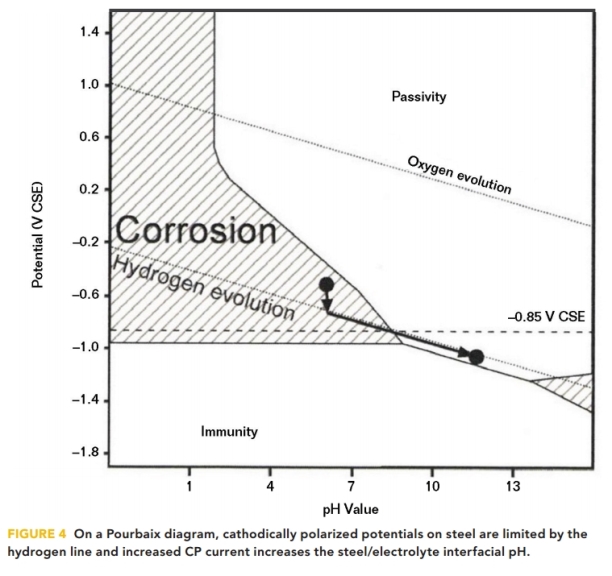
When CP current polarizes steel in the negative direction, the steel’s polarized potential normally cannot be forced more negative than 80 mV past the hydrogen line,9 where the electrolysis of water occurs and hydrogen gas is produced. Increasing the CP CD results in an increase in pH, where an increase of a single pH unit typically requires an order of magnitude increase in CP CD. When the structure’s polarized potential (Ep) resides on the hydrogen evolution line, the potential and the interfacial pH are linearly related by Equation (4):
Ep = –316 mV vs. CSE + (–59 mV x pH) (4)
The calculated potentials for pH 9 and pH 10 are –847 and –906 mV vs. CSE, respectively. It follows then that the structure’s polarized potential is an indirect indication of the interfacial pH when the structure polarized potential is at the hydrogen line.
In aqueous aerated solutions, the polarized potential of a structure may not be a linear relationship with pH because the structure/electrolyte potential does not meet the hydrogen line until the CP CD exceeds the oxygen-limiting CD (iL). This situation was illustrated by cathodic polarization scans carried out by Thompson and Barlo10 in simulated groundwater solutions, as shown in Figure 5.
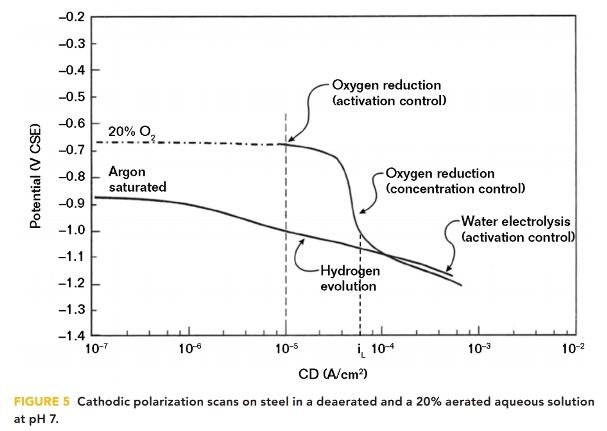
Argon saturation of the aqueous solution removed DO to obtain a deaerated condition where the structure/electrolyte potential is linearly related to the logarithm of applied CD. This is the hydrogen line, where water electrolysis and hydrogen evolution occur. It should be noted that to change the structure/electrolyte potential by 100 mV of cathodic polarization requires approximately a tenfold increase in the CP CD. Comparing these data with Figure 4 indicates there is a direct proportional relationship between pH and the logarithm of CD, which is similar to the potential/pH relationship on the hydrogen line.
For the 20% aerated condition, where 100% aeration would be ~8 ppm of DO, the structure’s polarized potential does not indicate the interfacial pH until the limiting CD for oxygen reduction is reached at a CD of ~10–4 A/cm2. The limiting CD increases as the DO concentration in the electrolyte increases.
Nevertheless, in an aerated solution, the charge transfer reaction is the reduction of DO and the production of OH– ions, even though the polarized potential is not linearly related to the pH. Even in an aqueous solution purged with air, the DO concentration at the structure/electrolyte interface is reduced to negligible concentrations as the structure is polarized to more negative values. This was demonstrated by Lewandowski, et al.11 in 1988 when they conducted cathodic polarization tests on stainless steel in a solution purged with air. At potentials more negative than –800 mV vs. CSE, the interfacial DO concentration was negligible. Where the interfacial environment is either naturally unaerated or deaerated because of the CP reduction reactions, a pH >9.5 is sufficient to reduce the corrosion rate to <25 μm/y (~1 mpy). When the interfacial environment is aerated, a pH >10.5 is sufficient to reduce the corrosion rate to <25 μm (~1 mpy).
The Interfacial pH Gradient
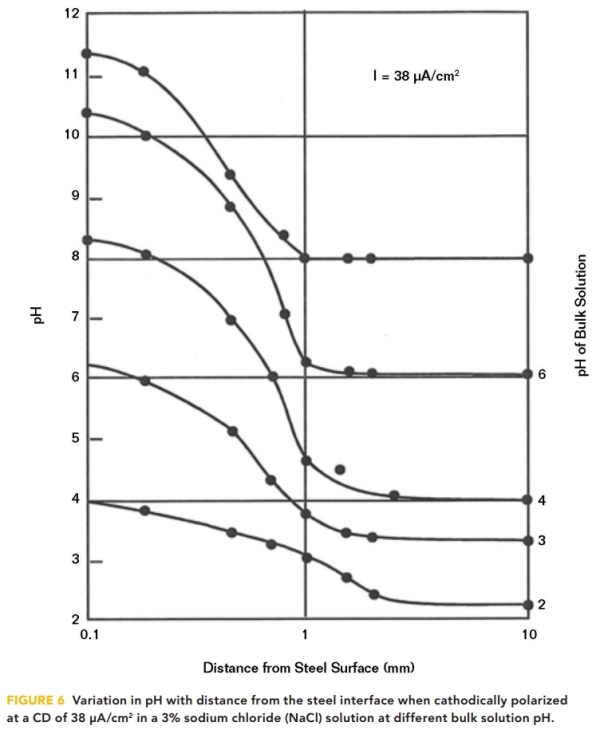
The highest pH occurs at the interface but diminishes rapidly as the distance from the interface increases, as shown in Figure 6. Measurements of pH at distances from the interface were taken by Kobayashi12 for various bulk solution pHs in aqueous solutions at a relatively high CD of 38 μA/cm2.
The solution pH generally drops to the bulk solution pH within a millimeter of the interface. This illustrates the difficulty in achieving protection on structures in a low pH solution. Even at a bulk solution pH of 4, the pH at the interface is only a little more than 8. To raise the interfacial pH by one unit would require increasing the CP CD by a factor of 10.
Although measuring the interfacial pH is not as easy as measuring the polarized potential, the interfacial pH is thermodynamically related to the polarized potential if the polarized potential resides at the hydrogen line of the Pourbaix diagram (e.g., when the structure/electrolyte interface is unaerated). Therefore, the polarized potential is an indirect indication of the interfacial pH, which in turn is an indication of the corrosion rate. The increase in pH, therefore, can be considered the predominant protection mechanism; and the polarized potential, except in aerated conditions, is simply an indication of the interfacial pH.
Steel Corrosion Potential vs. pH
It has been shown that the application of CP polarizes steel in the electronegative direction, while raising the pH at the interface at the same time. However, electronegative corrosion potentials can also be obtained in deaerated aqueous solutions simply by placing the steel in a high pH solution. This is predicted by the Pourbaix diagram for iron; but in aerated solutions, the steel corrosion potential is somewhat independent of pH, as shown in Figure 7.
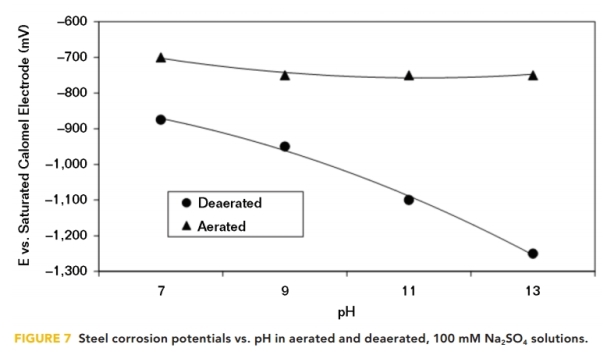
These corrosion potential data were produced as a result of cathodic polarization tests, conducted by Perdomo and Payer13 in 1995, on steel samples in a 100 mM sodium sulfate (Na2SO4) solution at varying pH values. They show that the corrosion potentials in the deaerated solutions were dependent on the pH, while in the aerated solutions they were virtually independent of the pH.
The CP mechanism could be interpreted as the increase in pH at the structure/electrolyte interface coupled with the consumption of DO in the oxygen reduction reaction, which creates a deaerated environment. Furthermore, in high resistivity, well-drained soils, both the NACE International and ISO CP standards14-15 permit less negative potential criteria than the –850 mV vs. CSE criterion, such as –750 and –650 mV vs. CSE. The pH in very aerated soils, however, can be very high at the interface despite these lower potentials, in which case the cathodic polarization mechanism can be attributed to the high pH developed at the interface. Therefore, the CP mechanism can be considered as “chemical polarization.”
Summary
• Increasing the pH at the steel/electrolyte interface decreases the corrosion rate in the absence of CP.
• The reduction reactions that transfer CP current from the electrolyte to the structure increase the pH and decrease the DO concentration in the electrolyte at the structure/electrolyte interface.
• In unaerated environments, the steel polarized potential is directly dependent on the interfacial pH produced by CP
• In aerated environments, the steel polarized potential is generally independent of the interfacial pH, which can be high even though the polarized potential is significantly less negative than the –850 mV vs. CSE criterion.
• In both unaerated and aerated conditions, the CP mechanism is arguably the development of a high pH caused by the operation of the CP system, which can be called “chemical polarization.”
References
1 R.B. Mears, R.H. Brown, “A Theory of Cathodic Protection,” Transactions of the Electrochemical Society 74 (1938): p. 527.
2 S.C. Dexter, L.N. Moettus, K.E. Lucas, “On the Mechanism of Cathodic Protection,” Corrosion 41, 10 (1985): p. 606.
3 R.J. Kuhn, “Cathodic Protection of Underground Pipe Lines from Soil Corrosion,” API Proc., Vol. 14, Section 4 (1933): p. 157.
4 W.J. Schwerdtfeger, O.N. McDorman, “Potential and Current Requirements for the Cathodic Protection of Steel in Soils,” Corrosion 11 (1952): p. 392.
5 T.J. Barlo, et al., “An Assessment of the Criteria for Cathodic Protection of Buried Pipelines,” AGA, Corrosion Supervisory Committee, June 1983, pp. 2-5.
6 N. Prakash, et al., “Corrosion of Mild Steel by Soil Containing Sulphate Reducing Bacteria,” J. Microbiological Biotechnology 3, 2 (1988): p. 82.
7 R.N. Parkins, R.R. Fessler, “Line Pipe Stress Corrosion Cracking—Mechanisms and Remedies,” CORROSION/86, paper no. 320 (Houston, TX: NACE International, 1986).
8 M. Büchler, “The Physical-Chemical Significance of the IR-Free Potential,” International Congress and Technical Exhibition (Brussels, Belgium: CECOR, 2013), p. 2.
9 S. Glasstone, An Introduction to Electrochemistry (Princeton, NJ: D. Van Nostrand Co., 1942), p. 443.
10 N.G. Thompson, T.J. Barlo, “Fundamental Processes of Cathodically Protecting Steel Pipelines,” Gas Research Conference Proc. held June 13-16, 1983 (Oslo, Norway: IGU, 1983).
11 Z. Lewandowski, et al., “Dissolved Oxygen and pH Microelectrode Measurements at Water Immersed Metal Surfaces,” CORROSION/88, paper no. 93 (Houston, TX: NACE, 1993).
12 T. Kobayashi, “Effect of Environmental Factors on the Protective Potential of Steel,” Proceedings of the 5th International Congress on Metallic Corrosion (Houston, TX: NACE, 1972), p. 628.
13 J.J. Perdomo, J.H. Payer, “Chemical and Electrochemical Conditions on Steel at Disbonded Coatings,” Pipeline Research Committee, AGA, Catalog # L51736, June 1995, pp. 109-119.
14 ISO 15589-1: 2015, “Petroleum and natural gas industries—Cathodic protection of pipeline systems—Part I: On-land pipeline (Geneva, Switzerland: ISO, 2015).
15 NACE SP0169-2013, “Control of External Corrosion on Underground and Submerged Metallic Piping Systems” (Houston, TX: NACE, 2013).
This article is based on CORROSION 2016 paper no. 7431, presented in Vancouver, British Columbia, Canada.